Article Text

Abstract
Objectives The objective of this study is the implementation of an automatic procedure to weekly detect new SARS-CoV-2 variants and non-neutral variants (variants of concern (VOC) and variants of interest (VOI)).
Methods We downloaded spike protein primary sequences from the public resource GISAID and we represented each sequence as k-mer counts. For each week since 1 July 2020, we evaluate if each sequence represents an anomaly based on a One Class support vector machine (SVM) classification algorithm trained on neutral protein sequences collected from February to June 2020.
Results We assess the ability of the One Class classifier to detect known VOC and VOI, such as Alpha, Delta or Omicron, ahead of their official classification by health authorities. In median, the classifier predicts a non-neutral variant as outlier 10 weeks before the official date of designation as VOC/VOI.
Discussion The identification of non-neutral variants during a pandemic usually relies on indicators available during time, such as changing population size of a variant. Automatic variant surveillance systems based on protein sequences can enhance the fast identification of variants of potential concern.
Conclusion Machine learning, and in particular One Class SVM classification, can support the detection of potentially VOC/VOI variants during an evolving pandemics.
- COVID-19
- machine learning
- public health informatics
- data science
Data availability statement
Data are available in a public, open access repository. Data are available in the GISAID repository: https://gisaid.org/.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Virus variants showing enhanced transmissibility, disease severity and other concerning characteristics arise during pandemics and they are usually detected by authorities after being isolated, and after their characteristics have emerged in a public health context.
WHAT THIS STUDY ADDS
We simulate an automatic variant surveillance system based on anomaly detection, able to detect a new variant as outlier based on its protein sequence.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Automatic variant surveillance systems can support the fast identification of new variants of concerns and variants of interests, thus prioritising interesting sequence variants for target laboratory testing.
Introduction
The ongoing pandemic caused by SARS-CoV-2 has seen the progressive emergence of different virus variants. The Centers for Disease Control and Prevention (CDC) has classified existing SARS-CoV-2 lineages into neutral variants, variants of interest (VOI) and variants of concern (VOC).1
VOI are variants with specific genetic markers that have been associated with receptor binding change, reduced neutralisation by antibodies and efficacy of treatments, potential diagnostic impact, predicted increase in transmissibility or disease severity. VOCs, on the other hand, are variants that, in addition to the possible attributes of a VOI, show impact on diagnostics, treatments or vaccines, interference with diagnostic test targets, substantially decreased susceptibility to therapies and neutralisation by antibodies, reduced vaccine-induced protection from severe disease, increased transmissibility or disease severity. A fourth classification, variants of high impact, is dedicated to variants more dangerous than VOCs, but none of the existing variants has been classified as such so far. Examples of VOCs are Alpha, Beta, Delta and Omicron.
Virus variants are classified after being isolated, and after their characteristics have emerged in a public health context, for example, enhanced transmissibility. For this reason, the countermeasures are always implemented after a variant is known, that is, the virus always has the upper hand in the arms race against the variants. Consequently, recognising a VOI or VOC as early as possible is utterly important to curb its damage, and ultimately save lives.
The virus protein sequences collected over the world are continuously deposited in the GISAID database, which was created in 2008 to promote influenza data sharing.2 GISAID is an example of informatics infrastructure implemented before the COVID-19 pandemic. It was of great importance to manage and monitor the COVID-19 emergence in the last 2 years.
Along with suitable strategies to collect and store the data, machine learning (ML) techniques have been extensively applied to analyse COVID-19 data.
Few studies are focused on variant-related predictions, for example, in isolating critical amino acid (AA) positions (or patterns) in the spike protein,3 or in forecasting novel variant potential waves.4 Importantly, these studies need input genomes that have already been isolated, that is, do not provide a viable method to generate novel genomes that could carry unknown but potentially dangerous variants. Of note, the Pango lineages framework has a specific ML module (PangoLearn).5 This module implements two simple ML approaches, decision trees and logistic regression, to classify unknown viral genomes into Pango lineages. The models are based on positional (alignment-dependent) features, and are limited to predicting known classes, that is, they can only predict known lineages.
To detect VOCs from their competitions with other variants, Zhao et al developed VOC-alarm, a statistical method based on the concept of mutational entropy.6 Authors defined the mutational entropy of a variant as a measure of the change of the mutation numbers across the globe for a lineage in a specific time period. In their analysis, Zhao et al noticed that some VOCs, such as Alpha, Delta and Omicron, grew from a small population and, as VOCs emerge, competing variants in precedent lineages decrease in population size.6 The concept of spreading mutations within a time window was also studied by Maher et al.7 A combined methodology was proposed by Makowski et al to evaluate single mutations in the spike proteins, based on two ML models, one to predict the impact of receptor binding domain mutations on ACE2 affinity and the other predicting human serum antibody affinity.8
Different from the aforementioned approaches, here we propose an ML method to timely predict the variants of concern as they are sequenced, without relying on information that needs to be collected over a period of time, such as changes in population size. That is, we develop an algorithm predicting each variant as being an ‘anomaly’ or not, using only the spike protein sequence, and ideally before the variants spread enough to manifest their related phenotypes—in other words ahead of their official classification. In recent work, we simulated the implementation of a pandemic surveillance classifier that predicts new non-neutral variants (VOCs and VOIs) monthly. Our system simulates a monthly update of a binary classifier with the new variants detected using supervised incremental learning.9 Incremental learning algorithms are able to incorporate new knowledge without a complete retraining of model parameters.10 For this reason, they can aid in evolving situations, such as during a pandemic. Yet, our incremental learning system assumes that the ground-truth class (neutral or non-neutral) for each variant is soon available at the end of the month. In the real case, this assumption does not always hold: for instance, the first Alpha sequence lately labelled as VOC was deposited in GISAID in late July 2020, while the Alpha variant was officially recognised as VOC by CDC only in late December of the same year.1
Here, we simulate the implementation of a pandemic surveillance classifier based on anomaly detection. Viruses continuously replicate, and during replications new types of variants that differ from the underlying population can arise. Detected anomalies can be new non-neutral variants. Briefly, we assume that we are in a peak state (in the space of spike protein sequences) when a specific variant is dominating the landscape, and the forthcoming of a new variant can be an anomaly that changes the state. Details of our proposed methodology can be found in the ‘Methods’ section. We will then evaluate the performance of our approach by comparing when our classifier predicts a known VOC/VOI as anomaly (in terms of date), with the date of designation as VOC by WHO as reported by the CDC. By predicting new virus sequences collected over time, the proposed approach can have the ability to raise a flag before to see variants are officially recognised as VOC/VOI by authorities.
Methods
Dataset
Our dataset consists of spike protein primary sequences from GISAID collected from February 2020 to March 2022. We decided to focus on spike protein sequences because VOC and VOI lineage classifications are based on mutations in spike proteins; moreover, by only focusing on the spike 1350 AAs, we limit the feature space (as opposed to considering all the SARS-CoV-2 proteins). After removing duplicated sequences, we filtered the spike proteins based on both the frequency of uncharacterised AAs, set to a maximum of 1%, and length, set to a minimum of 1000 AAs. From GISAID, we downloaded metadate with various information, such as variant type (‘unknown’, ‘Alpha’ and so on) and date of submission for each sequence.
Feature representation
We translate protein sequences into a fixed-length set of numeric features through k-mers, so that each protein, independently from its length, will have a numeric representation. K-mers are a classical method to represent biological nucleic or AA sequences, widely used in bioinformatics.11 Briefly, k-mers are substrings of user-defined length k contained in a sequence. For example, given k=2, we find in the sequence GATTACA the k-mers ‘GA’, ‘AT’, ‘TT’, ‘TA’ and ’CA’. Each k-mer has a Boolean value indicating its presence/absence. Since we wanted to represent variations of one to few AAs, we considered small ks, that is, k=3. We removed k-mers containing the ‘X’ character, indicating a missing value.
Variant surveillance implementation strategy
To simulate the implementation of the variant surveillance system, we hypothesised that in the last week of June 2020 a sufficient number of sequences that met our requirements were found in GISAID. In fact, 347 neutral variants were collected and we started the training process on these data. Moreover, to reduce the number of features (k-mers), we removed k-mers with zero counts in all the training sequences. This filtering step left us with 1922 k-mers. Subsequently, at each week, we collected the sequences from GISAID and the trained one class classifier predicted whether there are outliers (ie, non-neutral variants) among the new sequences. Starting from the predictions made each week, we calculate the confusion matrix weekly, where the negative samples (inlier) represent neutral variants, while positive samples (outlier) are VOC/VOI variants. Each week, it is evaluated whether in that week authorities recognised a new VOC or VOI, as reported in table 1. If so, the one class classifier is retrained on that week with all the sequences (1) in the initial training set and (2) the sequences that were predicted as inlier (ie, neutral) up until that week. A schematic representation of our training and test strategy is shown in figure 1. The classifier is developed using the implementation available in scikit-learn,12 in particular by using the One Class support vector machine (SVM). SVMs are early examples of supervised ML approaches applied to binary problems. To detect a possible non-linear decision boundary between two classes, SVM projects the data into a non-linear higher dimensional space by using a non-linear function. In such higher dimensional space, the data points belonging to the different classes are separated by a hyperplane that determines the margin between the two classes. One Class SVM is an adaptation of the binary SVM applied to novelty detection.13 In this case, the algorithm tries to separate the data points from the origin of the higher dimensional space. By doing so, the One Class SVM captures regions in the input space with different data density. One of the parameters that needs to be selected is the variable ν, that characterises the upper bound on the false positive (FP) fraction (training samples considered as outlier) and the lower bound on the number of training samples used as support vectors. In our implementation, we tested different combinations of SVM parameters. Based on the performance, from now on we will refer to the One Class SVM with non-linear kernel (radial basis functions) and low ν (0.01), which regulates the number of training samples that are allowed to be wrongly classified as outliers.
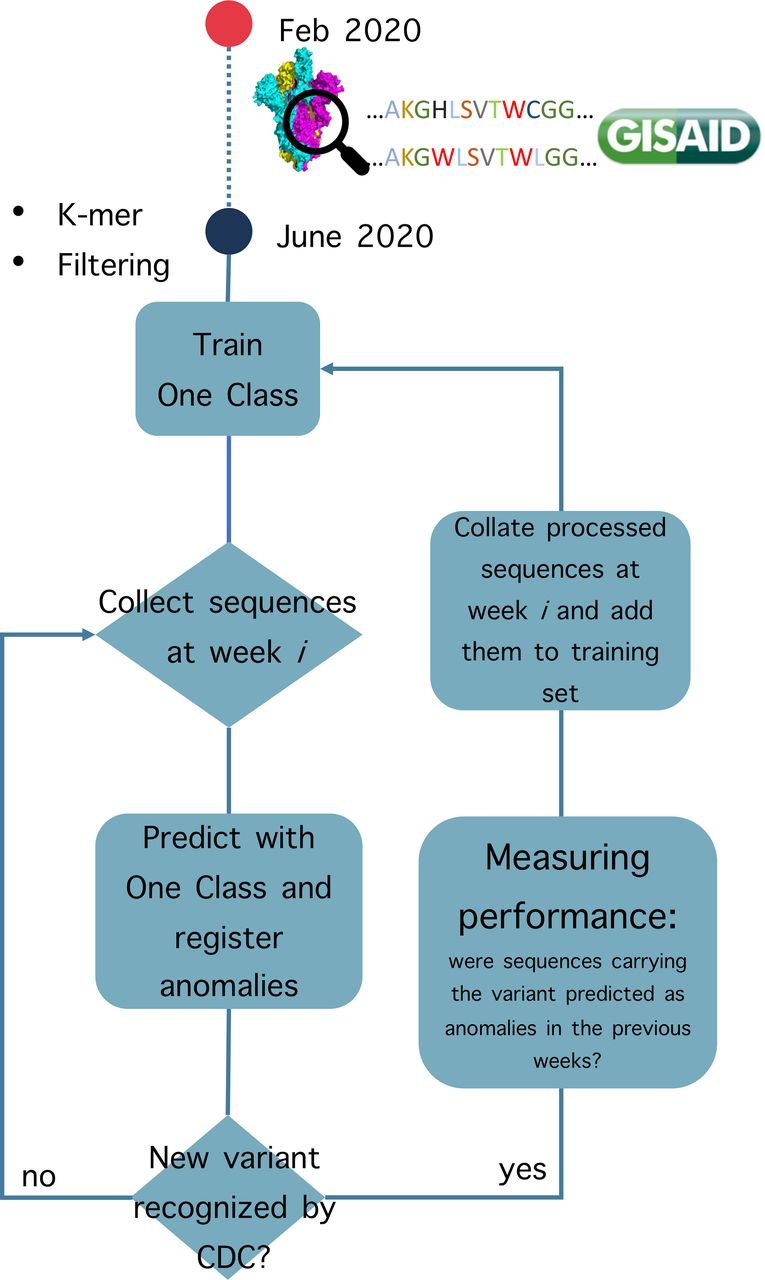
Schematic representation of automatic variant surveillance system. SARS-CoV-2 amino acid sequences deposited in GISAID from February 2020 to June 2020 are collected, transformed into k-mers and filtered. The anomaly detection system (One Class support vector machine (SVM)) is trained on this set of neutral variants. Then, at each following week, the newly uploaded sequences are predicted as either outlier or not. Predicted outliers are registered as anomalies. If authorities have recognised a new variants of concern (VOC)/variants of interest (VOI) in that week, the model is tested by evaluating whether the newly recognised VOC/VOI has already been predicted as an outlier by the One Class SVM in the previous weeks. CDC, Centers for Disease Control and Prevention.
List of VOCs and VOIs and their date of designation according to the CDC and WHO
To evaluate the model’s performance, we took into account the properties that a variant surveillance system should have to be useful in a realistic scenario. First of all, the problem can be highly imbalanced, and the imbalance rate varies across time (figure 2). Each week, new sequences to be predicted are made available. In a simulation of a real case scenario, each week we predict each newly sequenced sample, and the predicted outliers (ie, VOC/VOIs) are sent for further laboratory analysis that would eventually confirm whether or not each predicted-outlier variant is VOC/VOI. Since laboratory testing is time-expensive and costly, we would ideally send as few samples as possible to be analysed to reduce laboratory burden. For this reason, the cost of having a high number of false negatives (VOC/VOIs predicted as inliers) is lower than the cost of having a high number of FP (neutral variants predicted as outlier).

Number of neutral and non-neutral variants (VOC/VOI) for each week starting from 26 July 2020 to 23 March 2022. Red vertical lines indicate when a new variant was officially recognised as VOC/VOI according to the Centers for Disease Control and Prevention and WHO. VOC, variants of concern; VOI, variants of interest.
For these reasons, we evaluated our model in terms of the ability to detect at least one true VOC/VOI before the actual authority’s recognition and the number of predicted outliers. As far as performance metrics are concerned, we thus focus our attention on precision, which is calculated as the number of truly identified outliers (true positive (TP)) divided by the total number of predicted outliers (TP+FP).
Results
Dataset
Figure 2 shows the number of sequences collected, stratified by the class (neutral or VOC/VOI). For each VOI/VOC, the week containing the designation date by authorities is reported. As we can see, the number of sequences deposited in GISAID increased over time, starting from a few hundreds and reaching up to 10 000 in a week in late 2021. Moreover, while in the first year of pandemics the majority of sequences were neutrals, starting from March 2021 the number of non-neutral variants overtakes the number of neutral variants.
Automatic variant surveillance
Figure 3A and Figure 3B show the number and percentages of predicted outliers each week. As we can see, the percentage of predicted outliers varies, starting from 3.6% at the first week (17 predicted outliers out of 471 variants). The maximum number of predicted outliers occurs in week 60 (1095 predicted outliers out of 8201 sequences, 13.3%). The maximum percentage of predicted outliers is 21%, while the median value is 8%. Another important aspect to evaluate is the number of FP, that is, neutral variants that were incorrectly labelled as outliers, since a high number of FP will eventually increase laboratory burden. As we can observe from figure 3C, the number of FP is relatively low, with a maximum of 257 in week 35, corresponding to the 10% of the total number of neutral sequences analysed that week. In median, 9% of the neutral sequences are predicted as outliers (FP) each week. The ability to maintain low number of FP can be evaluated also from the precision (online supplemental table S1). As we can see from online supplemental table S1, the classifier initially strives to detect TP sequences, but as time passes the precision grows fast until it saturates towards >98%.
Supplemental material
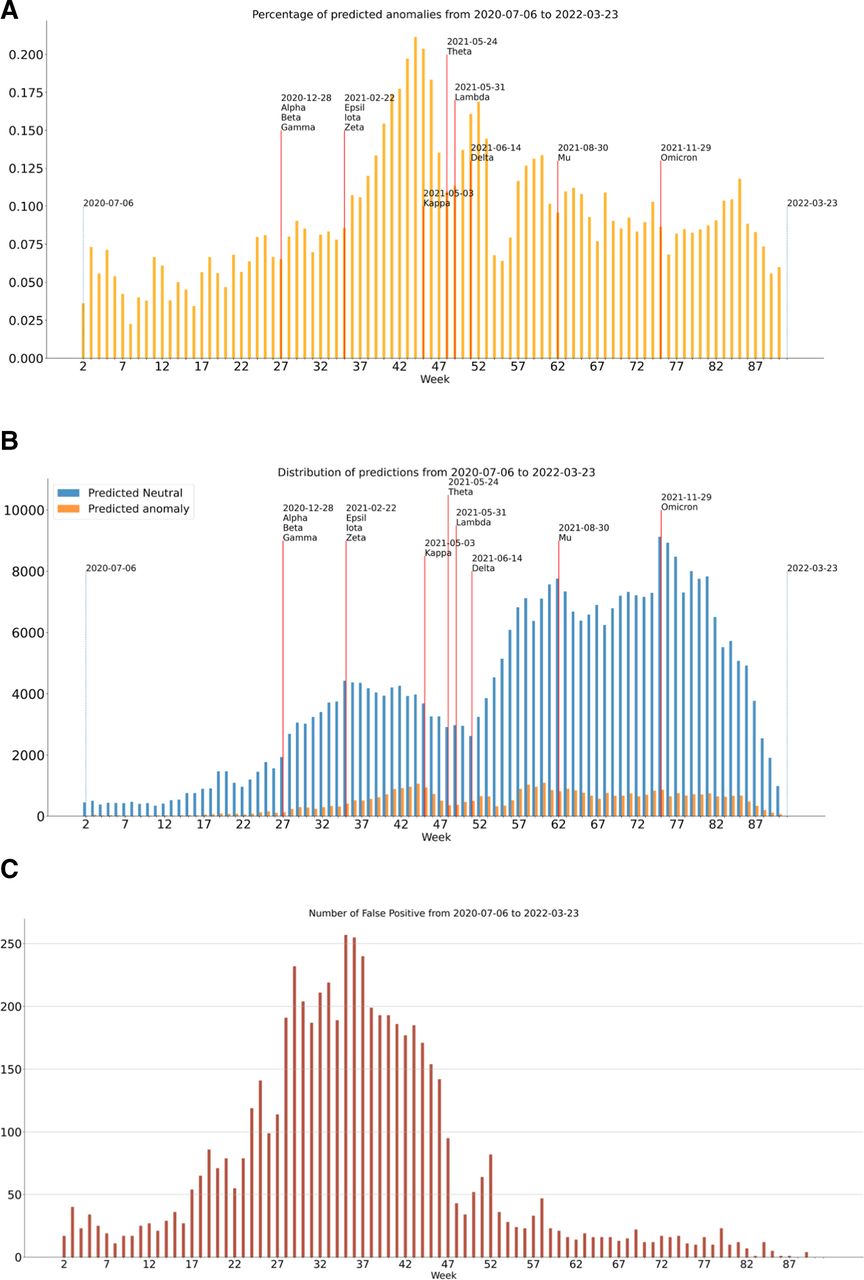
(A) Percentage of predicted outliers each week. (B) Number of predicted inlier and outlier each week. (C) Number of false positives, that is, neutral variants predicted as outliers.
Regarding the ability of our variant surveillance system to detect a new VOC/VOI as soon as possible, figure 4 reports, for each known variant type reported in GISAID data, the first time that the One Class classifier predicts at least one sequence of that type (red rhombus) and the actual time of designation by authorities (blue circle). As we can see, for all the variant types except for the Gamma, the classifier was able to detect at least a sequence of that type as outlier before the official designation. Gamma was recognised by the classifier in the same week of the official designation. The Alpha and Beta variants were recognised 8 and 7 weeks before, respectively. The Epsilon and the Kappa variants were detected 13 weeks before, while Iota was detected 6 weeks before. Zeta was identified 12 weeks before. Theta was recognised 8 weeks before. Lambda and Delta were detected 20 weeks before, Mu 21 weeks before and Omicron 3 weeks before. In median, a VOC/VOI was recognised as outlier 10 weeks before.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
For each variant type, the red rhombus indicates the first time that machine learning detects a variant as an outlier, while the blue circle indicates when the variant was officially recognised by authorities. For the Gamma variant, the week of detection by machine learning overlaps with the official recognition date. CDC, Centers for Disease Control and Prevention; VOC, variants of concern; VOI, variants of interest.
Discussion
During a pandemic caused by viruses such as the SARS-CoV-2, detecting new variants and understanding their effects as soon as possible is of paramount importance. Computational methods, such as ML, can highly support variant surveillance by uncovering patterns embodied in the huge amount of data that can be collected.14 We used the information encoded in spike protein sequences to spot anomalous variants. We show that our framework can be used in a real case scenario to select the most concerning variants (predicted as outlier by the anomaly detection system) for further laboratory testing to assess their potential harm even before they spread.
The development of ML tools for variant surveillance poses different challenges. First of all, a proper numerical representation of the protein AA sequences needs to be established. We chose to represent each protein with short k-mers. This simple representation proved to be effective in several proteomics and genomics problems.11 15 16 Unlike previous work,6 we only used patterns encoded in the AA sequences to predict non-neutral variants, without relying on information collected over time, such as changes in variant population size. As a result, our proposed system allows for the timely identification of non-neutral variants as soon as they are sequenced.
Second, the problem is highly imbalanced, and the number of sequences exponentially increases over time (figure 2). Additionally, the class composition varies: at the beginning, all deposited variants are neutral, while from 2021 the most competing variants, that is, VOCs and VOIs exceed the number of neutrals. This situation is a clear example of dataset shift, which often occurs in healthcare.17 18 Thus, a variant surveillance system needs to be able to adapt over time as the variant population changes. To do so, in a previous work we employed a binary incremental ML classifier, able to partially refit and consequently to update, the ML model over time.9 Yet, to achieve acceptable performance, we assume that the true class of variants (neutral vs non-neutral) was soon available at the end of each time step. To develop a more realistic system, here we propose to use One Class classification, in which the aim is to detect outliers, that is, instances that deviate from the normal population. Thus, we were able to train a classifier when zero non-neutral variants emerged, and the system identified deviations from the neutral population over time. To dynamically update the model, we decided to retrain the classifier when a new ground-truth classification was available, that is, when WHO officially recognises a new variant as VOC/VOI. At a given time step, the retraining is performed by using the initial training dataset plus the predicted inlier variants collected up until that time. This means that the classifier is retrained using also false negative variants, that is, VOC/VOI that were not predicted as outliers. As a matter of fact, using VOC/VOI as belonging to the inlier population does not affect the outcome of our procedure: we are not interested in predicting many (ie, the majority) of VOC/VOI as outliers, but we are interested in detecting few outliers that can be experimentally studied. Additionally, this retraining assumption allows the classifier to progressively predict less outliers for a given variant type that had emerged later in time, thus reducing the laboratory burden on variants that were already detected as outliers in the previous weeks. In fact, as we can see in online supplemental figures, the distribution of the predicted anomalies stratified by variant types showed that the number of predicted outliers is progressively decreasing after the peaks.
This work represents a proof-of-concept to show the feasibility of this apparently complex task with a simple feature representation (k-mers) and a solid ML algorithm (SVM). We recognised that other implementations, both for feature representation and prediction, can be applied to deal with this problem. For instance, deep learning, which is increasingly applied in a variety of fields, may be used in this case both for feature representation, through protein embedding,19 and as a predictive model for anomaly detection. In a recent work,20 authors analyse the features (in terms of mutations) of SARS-CoV-2 genomes, and map them on a Bayesian model to predict fitness. This approach can be complementary to our unsupervised model, which is focused on predictions, that is, through the spike protein, if a new genome carries a novel, unseen VOC/VOI. The work of Obermeyer et al, on the other hand, focuses on supervised interpretation of mutation important for the virus fitness, considering the whole genome, thus providing mechanistic insight. Future steps in our analysis can be inspired by this approach, for example, extracting k-mers (or k-mer modules) from the whole genome instead of only focusing on spike proteins, or using a supervised, white box approach to extract key features marking the making of novel VOC/VOI.
Conclusion
We have implemented an automatic variant surveillance system that exploits One Class classification to detect new potential VOC/VOI SARS-CoV-2 variants by evaluating the spike protein sequence. We evaluated the system ability to recognise a VOC/VOI as outlier before the official recognition by authorities. The classifier was able to detect a VOC/VOI with a median 10 weeks before, thus showing the potential utility of data-driven approaches to virus variant detection.
Data availability statement
Data are available in a public, open access repository. Data are available in the GISAID repository: https://gisaid.org/.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors GN carried on the experimental study and drafted the paper. SM contributed to the design and implementation of the experimental study and to the paper drafting. MS discussed the main ideas behind the research work and revised the manuscript. RB coordinated the research, contributed to the design of the experimental study and revised the manuscript. SM acceptes full responsibility for the work and had access to the data and controlled the decision to publish.
Funding This work was supported by EU Periscope Project grant number 101016233 and by NIH grant number R01 AI170187.
Competing interests GN is a full employee of Engenome s.r.l. RB is shareholder of Engenome s.r.l. and Biomeris s.r.l.
Provenance and peer review Commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.